{kind=link}
[3D .mol structure]

|
|
|
[3D .jpg image] [3D .mol structure] |
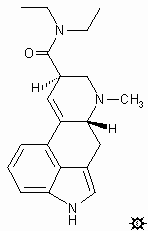
ALD-52. 1-Acetyl-N,N-diethyllysergamide. This material has been explored in the 50-175 microgram range and there are a number of human trials reported, with varying conclusions. One found that there was less visual distortion than with LSD and it seems to produce less anxiety and was somewhat less potent than LSD. Another report claimed it was more effective in increasing blood pressure. Yet another could not tell them apart. ALD-52 just may have been the drug that was sold as "Orange Sunshine" during the "Summer of Love" in the late '60's. Or "Orange Sunshine" may have been, really, LSD. This was the focus of a fascinating trial where two defendants were accused of distributing LSD, whereas they claimed that it was ALD-52 which was not an illegal drug. The prosecution claimed that as it hydrolyses readily to LSD, for all intents and purposes it was LSD, and anyway, you had to go through the illegal LSD to get to ALD-52 by any of the known chemical syntheses. The defendants were found guilty. And yet, I do not know who has actually measured the speed or ease of that reaction. If ALD-52 hydrolyses so easily to LSD, and the body is indeed a hydrolytic instrument, then these two drugs should be absolutely equivalent in every particular, This is the ergot equivalent of the psilocybin to psilocin argument, except this is an acetamide rather than a phosphate ester.
--indole-ring substituent-- at N-1 at C-2 code -H
-COCH3
-CH3
-CH2OH
-CH2N(CH3)2
-H
-H
-CH3
-CH3-H
-H
-H
-H
-H
-Br
-I
-Br
-ILSD-25
ALD-52
MLD-41
BOL-148
MBL-61
MIL
| indole | --- amide nitrogen substituents --- | ||
| R= | R= | R= | code name |
| -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -H -CH3 -CH3 -COCH3 -CH3 |
-H -CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)CH2OH -CH(CH3)CH2OH -(CH2)CH3 -CH(CH3)CH2CH2OH -(CH2)4CH3 -CH(CH3)CH2CH2CH3 -CH(CH2CH3)2 -(CH2)5CH3 -CH(CH3)CH2CH2CH2CH3 -(CH2)6CH3 -CH(CH3)CH2CH2CH2CH2CH3 -CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)2 -CH(CH3)CH2C6H5 -CH2CH3 -(CH2)2CH3 -(CH2)2CH3 -CH(CH3)2 -CH2CH=CH2 -(CH2)2CH3 -CH2CH=CHCH2- -CH2CH2CH2CH2CH2- -CH2CH2OCH2CH2- -CH(CH2CH3)CH2OH -COCH3 |
-H -H -H -H -H* -H -H* -H* -H -H* -H -H -H* -H -H* -CH3 -CH3 -CH3 -CH3 -CH3* -CH2CH3 -CH2CH3 -(CH2)2CH3 -CH(CH3)2 -CH2CH=CH2 -(CH2)3CH3 -H -H* -CH2CH3 |
LA-111 LAE-32 Ergonovine Methergine DAM-57 LAMP LSD-25 DAL LPD-824 LSM-775 MLA-74 UML-491 ALA-10 MPD-75 |
| [ |
[Main Index] | [Forward |
| HTML and Design by Bo & Erowid | Used by Erowid with permission of author |
|
[Plants & Drugs]
[Mind & Spirit]
[Freedom & Law]
[Arts & Sciences]
[Library]
[Search]
[About] (html and design © 1995-2009 Erowid.org. Please ask permission before publicly reproducing.) (Contents © respective copyright holders.) |
|---|