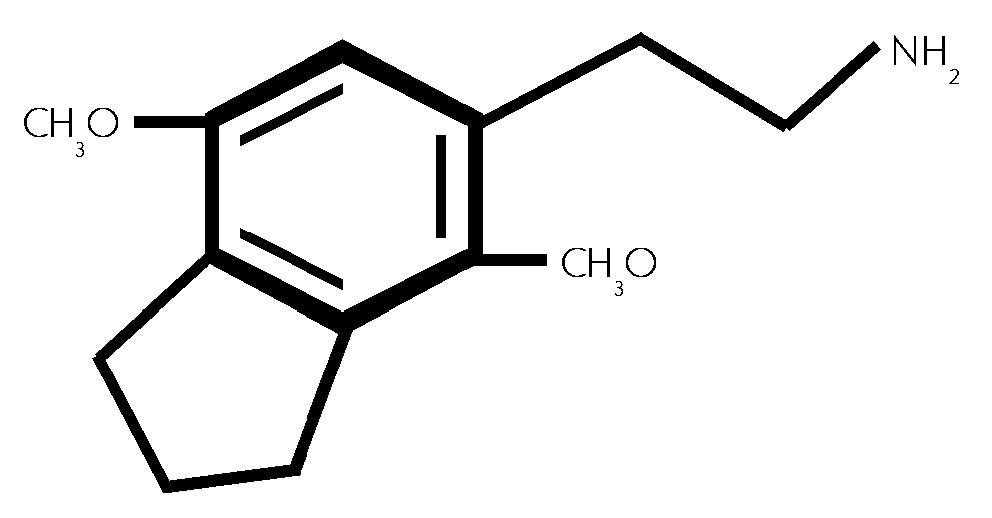
(#42/527)
2,5-Dimethoxy -3,4- (trimethylen) -phenethylamin, 5- (2-Aminoethyl) -4,7- dimethoxyindan. (#42/526)
Dieses Halluzinogen ist von der chemischen Struktur her ein Phenethylamin. Es ist damit nahe verwandt mit den Halluzinogenen 2C-B, 2C-E, Meskalin, oder auch 2C-C. Die einzelnen Phenethylamine unterscheiden sich nur durch die Art der Substituenten am aromatischen Benzolring. (eigen)
Haengt man bei einem Phenethylamin noch eine Methylgruppe an der Ethylaminseitengruppe an, kommt man zu der Gruppe der Amphetamine. Grundsaetzlich scheint die Regel zu gelten, dass wenn ein Phenethylamin psychoaktiv ist, ist auch sein Amphetamingegenstueck wirksam ist. Mehr noch, es ist sogar allgemein staerker. (eigen)
Alexander Shulgin gibt in seinem Buch "PIHKAL" noch folgende zusaetzliche Kommentare ueber die Natur des Halluzinogens:
Das Positive eines ganz faszinierenden, veraenderten Bewusstseinszustandes, frei von offensichlichen physischen Bedrohungen, ist hier mit der Negativitaet der langen Wirkdauer gepaart. Es gibt eine heitere Verruecktheit, die eine freudige Vergiftung ergeben kann, aber mit der darunter liegenden Paranoia, wie es andere sehen. Es gibt an Einfachheit der Kommunikation, aber nur in Umgebungen, die wohlbekant und freundlich sind. Dies koennte eine wirklich beaengstigende Erfahrung sein, wenn sie sich in unfamiliaerer oder unstrukturierter Umgebung ereignet. (#42/529)
Von diesem Halluzinogen existieren subjektive Wirkungsbeschreibungen, die von A. Shulgin in seinem Buch "PIHKAL" angegeben werden:
16mg: Es setzte in kleinen Spruengen ein. Alles beruhigte sich, und dann nahm es einen zusaetzlichen kleinen Sprung aufwaerts. Ich bin ganz konzentriert. Schreiben ist einfach. Mein Appetit ist maessig. Wuerde ich in die Stadt fahren, um ein Buch zurueckzugeben? Kein immer geliebter Weg! Ich bin sehr zufrieden, gerade hier zu sein, wo ich sicher bin, und bleibe beim Schreiben. Es braucht so viel Zeit zu sagen, was gesagt werden will, aber es gibt keinen schnellen Weg. (#42/529)
22mg: Ich ging hinaus fuer die Post genau zur Daemmerung. Dies war das tapferste Ding, dass ich moeglicherweise tun konnte, gerade fuer eine lausige Postkarte und ein Journal. Was waere, wenn ich jemanden getroffen haette, der reden wollte? Gegen abend bekam ich einen Anruf von Peg, die sagte, dass ihre Bohnensuppe unheimlich brodelte und was sie tun sollte, und ich sagte, es waere vielleicht besser Suppe zu machen. Es war diese Art von Erfahrung! Mengen an LSD-aehnlichen Funken, und nichts macht wirklich Sinn. Wunderbar. (#42/529)
25mg: Es war einfach zu sprechen, und kein Hinweis auf Koerperwirkungen. Der Schlaf an diesem abend war einfach. Der naechste Tag war voll guter Energie. (#42/529)
Alexander Shulgin gibt in seinem Buch "PIHKAL", dem Standardwerk ueber halluzinogene Phenethylamine und Amphetamine, eine Dosis von 16-25mg an, indem die Droge halluzinogen wirkt. (#42/528)
Die Dauer der halluzinogenen Wirkung wird von A. Shulgin mit 12-24h angegeben. (#42/529)
Die Ausbildung einer koerperlichen Abhaengigkeit ist nicht bekannt. (eigen)
A. Shulgin gibt in seinem Buch "PIHKAL" folgende Synthese fuer das Halluzinogen 2C-G3 an:
Zu einer Loesung von 22g KOH in 250ml heissem EtOH wurden 50g 4-Indanol und 75g Methyliodid hinzugefuegt. Die Mischung wurde 12h lang am Rueckfluss gehalten. Es wurden dann zusaetzliche 22g KOH hinzugefuegt, gefolgt von zusaetzlichen 50g Methyliodid. Kochen am Rueckfluss wurde zusaetzliche 12h fortgesetzt. Die Mischung wurde in 1l H2O gegossen, mit HCl angesaeuert, und mit 3x75ml CH2Cl2 extrahiert. Die vereinten Extrakte wurden mit 5%-iger NaOH gewaschen, dann mit verd. HCl. Das Loesungsmittel wurde im Vakuum entfernt. Der Rest des rohen 2,3-(Trimethylen)anisol wog 56.5g und wurde ohne weitere Reinigung in der folgenden Reaktion gebraucht.
Einer Mischung von 327g N-Methylformanilid und 295g POCl3 wurde es erlaubt sich im Inkubator zu entwickeln, bis sich eine dunkelweinrote Farbe gebildet hatte. Es wurde dann 110g rohes 2,3-(Trimethylen)anisol hinzugefuegt. Die Mischung wurde am Dampfbad erhitzt. Es gab eine kraeftige Entwicklung von Gasen, welche sich grossteils nach etwas 4h Erhitzen beruhigte. Die Reaktionsmischung wurde zu 4l H2O hinzugefuegt und die Nacht lang geruehrt. Die oelige, waessrige Phase wurde mit 3x200ml CH2Cl2 extrahiert. Nach Vereinen der Extrakte und dem Entfernen des Loesungsmittels wurden 147g schwarzes, suessriechendes Oel erhalten. Dieses wurde bei 182-194 Grad Celsius an der Wasserstrahlpumpe destilliert, um 109.1g blass gelbes Oel als Ausbeute zu erhalten. Bei niederer Temperatur kristallisierte dieses, aber die Feststoffe schmolzen wieder bei Zimmertemperatur. Gaschromatographie dieses Produkts auf OV-17 bei 185 Grad Celsius zeigte feststellbares anfaengliches Anisol und N-Methylformanilid (vereint, vielleicht 5% des Produkts) und eine kleine aber existente isomerische Spitze, (rund 5%, leicht schneller bewegend als das Aldehyd der Ueberschrift, wieder rund 5% des Produkts), von der versuchsweise das ortho-Aldehyd (2-Methoxy-3,4-(trimethylen)-benzaldehyd) identifiziert wurde. Die Masse dieses Rohprodukts (74g) wurde wieder bei 110-130 Grad Celsius mit einem Druck von 0.3mm/Hg destilliert, um 66g 4-Methoxy-2,3-(trimethylen)benzaldehyd zu ergeben, als beinahe farbloses Oel, welches einen kristallinen Feststoff bildete. Eine Portion auf einer poroesen Platte zeigte einen mp von 28-29 C. 1g von diesem Aldehyd und 1g Malononitril in 25ml EtOH wurde mit einigen wenigen Tropfen Triethylamin versetzt und ergab blass gelbe Kristalle des Malononitrilderivats. Dieses, nach dem Umkristallisieren aus 50ml kochendem EtOH, hatte einen mp von 176-176.5 Grad Celsius.
Eine Loesung von 34.8g 4-Methoxy-2,3-(trimethylen)benzaldehyd in 800 ml CH2Cl2 wurde mit 58.6g 85%-iger m-Chlorperoxybenzoesaeure versetzt und wurde 3 Tage lang am Rueckfluss gehalten. Nach Abkuehlen und einigen Tage Ruhen wurden die Feststoffe durch Filtration entfernt und sparsam mit CH2Cl2 gewaschen. Die vereinten Filtrate und Waschungen wurden mit 200ml gesaettigter NaHCO3-Loesung gewaschen. Das Loesungsmittel wurde entfernt, was 43.5g dunkel gefaerbtes Oel ergab. Dieses wurde in 150 ml MeOH geloest, zu dem 9g NaOH hinzugefuegt worden waren. Alles wurde am Rueckfluss auf dem Dampfbad erhitzt. Nach 1h wurde eine Loesung von 9g NaOH in 20ml H2O hinzugefuegt, weiter erhitzt, dann noch eine weitere Behandlung mit 9g NaOH in 20ml H2O, gefolgt von zusaetzlichem Erhitzen. Alles wurde zu 800ml H2O hinzugefuegt, einmal mit CH2Cl2 gewaschen (was eine geringfuegige Menge des Materials entfernte) und dann mit HCl angesaeuert. Die dunklen Kristalle, die sich bildeten, wurden filtriert und an der Luft zu einem konstantem Gewicht getrocknet, was 27.5g dunkle, aber nett aussehende Kristalle ergab, mit einem mp von 89-91 Grad Celsius.
Zu einer Loesung von 10g KOH in 100g EtOH, die 5% IPA beinhalteten, wurden 27.5g von den obigen bei 89-91 Grad Celsius schmelzendem Material hinzugefuegt, gefolgt von 25g Methyliodid. Die Mischung wurde uebernacht am Rueckfluss gehalten. Alles wurde zu 800ml H2O hinzugefuegt, mit HCl angesaeuert, und mit 3x100ml CH2Cl2 extrahiert. Die vereinten Extrakte wurden mit 3x100ml 5%-iger NaOH gewaschen, dann einmal mit verd. HCl. Das Loesungsmittel wurde im Vakuum entfernt, was 20.4g wohlriechenden, kristallinen Rest ergab. Dieser wurde aus 60ml kochendem MeOH umkristallisiert, um 16g 1,4-Dimethoxy-2,3-(trimethylen)benzol (4,7-dimethoxyindan) zu ergeben, nach Filtration und Lufttrocknen, mit einem mp von 86-88 Grad Celsius.
Zu einer Mischung von 39g N-Methylformanilid und 35.9g POCl3, der es erlaubt worden war bei Umgebungstemperaturen zu stehen, bis sie dunkelweinrot war (rund 45 Min.), wurden 15.8g 1,4-Dimethoxy-2,3-(trimethylen)benzol hinzugefuegt. Die Mischung wurde auf dem Dampfbad 4h lang erhitzt. Dann wurde sie in 600ml H2O gegossen. Nach einer Nacht Ruehren wurde eine schwere kristalline Masse erzeugt. Diese wurde durch Filtration entfernt. Nach dem Lufttrocknen, wurde sie mit 3x100ml kochendem Hexan extrahiert. Vereinen und Kuehlen dieser Extrakte ergab 9.7g lachsfarbene Kristalle mit einem mp von 67-68 Grad Celsius. Diese wurden aus 25ml kochendem EtOH umkristallisiert, um 7.4g 2,5-Dimethoxy-3,4-(trimethylen)benzaldehyd zu ergeben, nach Filtration, EtOH Waschen und Lufttrocknen bis zu konstantem Gewicht, mit einem mp von 71-72 Grad Celsius. Die Mutterfluessigkeiten ergaben bei vorsichter Behandlung mit H2O, nach Umkristallisieren aus EtOH, 1g zusaetzliches Produkt.
Eine Loesung von 150mg Aldehyd und dem gleichen Gewicht an Malononitril in 2.3ml EtOH, mit 3 Tropfen Triethylamin versetzt, ergab sofort gelbe Kristalle des Malononitrilderivats, mit einem mp von 161-162 Grad Celsius.
Eine Loesung von 3.7g 2,5-Dimethoxy-3,4-(trimethylen)benzaldehyd in 15g Nitromethan wurde mit 0.7g wasserfreiem Ammoniumacetat versetzt und auf dem Dampfbad 14h lang erhitzt. Die fluechtigen Stoffe wurden im Vakuum entfernt. Der Rest bildete 3.5g dunkle Kristalle, die breitgefaechert zwischen 126-138 Grad Celsius schmolzen. Umkristallisieren der gesamten Masse aus 70ml kochendem EtOH ergab 3.2g glaenzend goldene Kristalle mit einem mp von 129-137 Grad Celsius. Eine weitere Umkristallisierung eines analytischen Beispiels aus MeOH ergab 2,5-Dimethoxy-3,4-(trimethylen)-beta-nitrostyren, als gelbe Kristalle mit einem mp von 146-147 Grad Celsius.
Zu einer kalten Loesung von LAH in THF (40ml einer 1M Loesung), gut geruehrt und unter einer Inertatmosphaere, wurde tropfenweise 1.05ml frisch bereitete 100%-ige H2SO4 hinzugefuegt. Es wurden dann tropfenweise eine Loesung von 2.39g 2,5-Dimethoxy-3,4-(trimethylen)-beta-nitrostyren in 25ml THF hinzugefuegt. Die hellgelbe Farbe wurde sofort ausgebleicht. Nachdem die Addition fertig war, wurde das Ruehren fuer zusaetzliche 20 Min. fortgesetzt. Die Reaktionsmischung wurde zum Kochen am Rueckfluss am Dampfbad fuer zusaetzliche 0.5h gebracht. Nach dem Abkuehlen wurde das ueberschuessige Hydrid mit IPA (8ml verwendet) zerstoert, gefolgt von genuegend 15%-iger NaOH, um die anorganischen Stoffe in eine lockere, filtrierbare Masse zu wandeln. Diese wurden durch Filtration entfernt. Der Filterkuchen wurde mit THF gewaschen. Die vereinigten Filtrate und Waschungen wurden vom Loesungsmittel im Vakuum befreit, und der Rest in verduennter H2SO4 geloest. Nach Waschen mit CH2Cl2 wurde die waessrige Phase mit 25%-iger NaOH basisch gemacht und mit 3x75ml CH2Cl2 extrahiert. Nach Entfernen des Loesungsmittel im Vakuum wurde der Rest bei 125-160 Grad Celsius mit einem Druck von 0.45mm/Hg destilliert, um 0.8g weisses Oel an Ausbeute zu ergeben. Dies wurde in 8ml IPA geloest, mit 20 Tropfen konz. HCl neutralisiert (die Salzkristalle begannen sich zu bilden, bevor dies fertig war) gefolgt von der Addition von 65ml wasserfreiem Et2O. Die weisse kristalline Masse wurde filtriert, mit Et2O gewaschen und an der Luft getrocknet, um 1.16g 2C-G-3-HCl, mit einem mp von 214-216 Grad Celsius bei Zersetzung, zu erhalten. Anal. (C13H20ClNO2) C,H. (#42/526ff.)