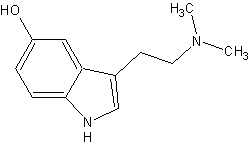
(#101)
Bufotenin; 5-HO-DMT; 5-Hydroxy-dimethyltryptamin. (eigen)
TRYPTAMINE, 5-HYDROXY-N,N-DIMETHYL; INDOL-5-OL, 3-[2-(DIMETHYLAMINO)ETHYL]; 5-HYDROXY-N,N-DIMETHYLTRYPTAMINE; 3-(2-DIMETHYLAMINOETHYL)INDOL-5-OL; N,N-DIMETHYLSEROTONIN; BUFOTENINE; MAPPINE; (engl.) (#101)
3-[2-(Dimethylamino)ethyl]-1H-indol-5-ol; 3-(2-dimethylaminoethyl)-5-indolol; 5-hydroxy-N,N-dimethyltryptamine; N,N-dimethylserotonin; 3-(N-dimethylaminoethyl)-5-hydroxyindole; mappine; (MerckIndex, 13. Aufl.)
Freebase Bufotenin
CAS Registry Nummer: 487-93-4(MerckIndex, 13. Aufl.)
Summenformel: C12H16N2O; (MerckIndex, 13. Aufl.)
Mol.gewicht: 204.27 g/mol. (MerckIndex, 13. Aufl.)
Zusammensetzung: C 70.56%, H 7.89%, N 13.71%, O 7.83%; (MerckIndex, 13. Aufl.)
Schmelzpunkt: Stout Prismen aus Ethylacetat; 146147 Grad Celsius (295297 Grad Fahrenheit) (MerckIndex, 13. Aufl.)
Siedepunkt:0.1: 320 Grad Celsius (608 Grad Fahrenheit); (MerckIndex, 13. Aufl.)
Starke Prismen aus Ethylacetat. (MerckIndex, 13. Aufl.)
Opt. Aktivitaet: uv max: 220, 265 nm (log * 4.0, 3.7). (MerckIndex, 13. Aufl.)
Beinahe unloeslich in Wasser. Frei loeslich in Alkohol, weniger loeslich in Aether. Loeslich in verd. Saeuren und Alkalien. (MerckIndex, 13. Aufl.)
XLogP: 1.6;
XLogP3: 1.2; (PubChem)
pKa:
Loeslichkeit:
Aceton 20 C: loeslich (5 g/100 ml); (annonym: Internet)
Chloroform 20 C: loeslich; (annonym: Internet)
Dichlormethan 20 C: loeslich; (annonym: Internet)
Dimethylsulfoxid (DMSO) 20 C: loeslich (6 g/100 ml); (annonym: Internet)
D-Limonen (Orangen Oel) 20 C: unloeslich; (annonym: Internet)
D-Limonen (Orangen Oel) 176 C: loeslich (mehr als 1.7 g/100 ml); (urspruenglich annonym geposted, von DMT-Nexus zur Diskussion und zur oeffentlichen Quellenueberpruefung uebernommen)
Verd. Saeuren und Basen: loeslich (Merck Index); (annonym: Internet)
Ethanol 20 C: loeslich; (annonym: Internet)
Ether 20 C: loeslich; (annonym: Internet)
Ethylacetat 20 C: loeslich; (annonym: Internet)
Heptan 20 C: unloeslich; (annonym: Internet)
Heptan mit 40% MEK 20 C: loeslich (0.53 g/100 ml); (annonym: Internet)
Heptan mit 50% MEK 20 C: loeslich (1.22 g/100 ml); (annonym: Internet)
IPA 20 C: loeslich; (annonym: Internet)
MEK 20 C: loeslich; (annonym: Internet)
Methanol 20 C: loeslich; (annonym: Internet)
Naphtha 20 C: unloeslich; (annonym: Internet)
Wasser 20 C: beinahe unloeslich in reinem Wssser (keine Seaure oder Base zugefuegt); (annonym: Internet)
Xylen 20 C: beinahe unloeslich (weniger als 0.03 g/100 ml); (annonym: Internet)
Xylen 144 C: loeslich (1.5 g/100 ml);(annonym_ Internet, von DMT-Nexus zur Diskussion und zur oeffentlichen Quellenueberpruefung uebernommen)
Bufotenin Fumarat
Loeslichkeit:
loeslich in Wasser
unloeslich in FASA (Fumarsaeure gesaettigtes Aceton); (DMT-Nexus)
Bufotenin Methyliodid:
Summenformel: C13H19IN2O;
Mol.gewicht: 346.21;
Zusammensetzung: C 45.10%, H 5.53%, I 36.66%, N 8.09%, O 4.62%;
Eigenschaften: Starke Prismen aus Methanol, 214-215 Grad Celsius erniedrigter mp. (MerckIndex, 13. Aufl.)
Bufotenin ist ein Tryptamin von der chemischen Struktur her und damit nahe verwandt mit einer Reihe pharmazeutisch aktiver Substanzen. Es hat jedoch nicht die starke halluzinogene Komponente, die fuer seine Verwandten DMT, DET oder Psilocybin typisch ist, vielmehr sollen unangenehme koerperliche Nebenwirkungen ueberwiegen, wurde bahuptet, anhand von Versuchen mit injiziertem Bufotenin. Dennoch wurde Bufotenin oftmals in der Literatur als Halluzinogen beschrieben, was wir auch selbst festgestellt haben, es stimmt. Bufotenin hat eine weite Verbreitung im Tier- und Pflanzenreich. Es kommt in verschiedenen Kroetenarten vor, ebenso aber auch in verschiedensten Pflanzen, wo es oftmals als Begleiter bekannter halluzinogener Tryptamine auftritt und fuer visuelle Nebenwirkungen der Pflanzendrogen verantwortlich ist. Es ist in der Anadenanthera colubrina als wichtigstes Alkaloid, neben Spuren von DMT enthalten, und ist mitveranwortlich fuer die Gesamtwirkung der yopo und coboha oder auch vilca und cebil-Schnupfpulver der suedamerikanischen Konsumenten, in vergangenen Kulturen (auf den Westnindischen Inseln ist es, scheint so, - ausgestorben) und auch noch heute konsumiert wird. Der Schnupfpulvergebrauch schien schon teilweise ausgestorben, bis er von findigen Menschen wiederbelebt wurde. So ist es heute problemlos moeglich ueber einschlaegige Firmen via Internet die Samen zu beziehen und das originale Rezept der Schnupfpulverbereitung nachzuvollziehen. Dies wurde auch von uns gemacht und wir nahmen nicht den beiliegenden geloeschten Kalk, der sehr aggresiv in der Nase sein wuerde, wenn man ihn nicht richtig verduennt, sondern normale Asche, wie es die Indianer ehedem auch getan haben. Wir haben staerkste Wirkungen durch dieses Schnupfpulver festgestellt, aber auch Uebelkeit am Anfang und Erbrechen, bis wir in die Erfahrung durchgebrochen waren. Nach diesen harten Anfangszeiten haben wir mehr als nur die volle Palette des DMT-Trips erfahren. Das wenige DMT alleine kann gar nicht so stark sein, sondern das viele in den Samen enthaltene Bufotenin verursacht die Wirkung, da es selbst eindeutig halluzinogen ist. Was soll es denn sonst sein, es wurde nicht anderes in nennenswerten Mengen festgestellt. Bufotenin ist eindeutig halluzinogen und ist mitverantwortlich fuer die Wirkungen der obenangefuehrten Schnupfpulver. (eigen)
Anschliessend noch ein email-posting zum Thema Bufotenin, das die wesentlichen Neuerungen kurz zusammenfasst:
Bufotenin ist in Anadenanthera colubrina Samen enthalten und es ist sehr einfach zu extrahieren (braucht aber eine unterschiedliche Prozedur wie DMT). Bufotenin sollte nicht oral eingenommen oder injeziert werden. Es ist unangenehm, wenn man es ueber diese Route einnimmt. Aber es ist sehr nett, wenn man es raucht. (dmt-nexus, Vlad, Dienstag, 7. Juli 2009 00:02:36)
Aufgrund fehlender eigener Erfahrung mit reinem Bufotenin, soll kurz ein Ausblick auf die internationale, forschende Gemeinschaft gegeben werden, die sich mit der Wirkung von Bufotenin intensiv auseinandersetzt:
Eine bessere Rauch-Kombination ist Harmin-freebase mit Bufotenin-freebase, welche angenehme Visionen in den meisten Konsumenten erzeugt, wurde berichtet, weil der trip primaer visuell ist, ohne Ichverlust und andere erschreckende, psychedelische Wirkungen von DMT. Es ist nicht so fremdartig wie DMT, viel mehr visuelles, und dauert auch laenger (2-3 Stunden). (dmt-nexus, Vlad, Dienstag, 7. Juli 2009 00:02:36)
Am Amazonas ist es ueblich Caapi Winde zu kauen, waehrend man Vilca (ein Schnupfpulver bereitet aus Anadenanthera colubrina) schnupft, um eine kurze ayahuasca-aehnliche Erahrung zu produzieren. Es ist nicht so freaky wie ayahuasca, aber es ist mehr visuell. Bufotenin ist auch mehr visuell als DMT, aber es ist nicht so psychedelisch wie DMT. Alle schlechten Informationen, die Leute gehoert haben ueber Bufotenin, kommen davon, dass es injeziert wurde. Es ist sehr giftig und unangenehm wenn es injeziert wird, aber nicht wenn es geraucht wird. (dmt-nexus, Vlad, Dienstag, 7. Juli 2009 00:02:36)
Bufotenin (5-HO-DMT) verursacht keine Uebelkeit. Es ist sehr nahe verwandt zu Psilocin (4-HO-DMT). Das Visuelle ist in der Art von Psilocin. Es ist ein sehr freundliches Psychedelika, einfacher zu handhaben als Psilocin, und viel einfacher als DMT. Es erzeugt eine Menge auditorischer Halluzinationen. Mehr als irgendein anderes Halluzinogen, von dem ich gehoert habe. Es ist auch sehr euphorisch. Das nette daran ist, verglichen mit anderen Halluzinogen ist, dass es ist sehr intensiv visuell und auditorisch ist, ohne viel andere Wirkungen zu erzeugen, auch mit hohen Dosen. Wirkungen wie Ichverlust, Paranoia, Zeitverlangsamung, und andere eigenartige halluzinogene Wirkungen, bekannt von LSD, DMT, etc., existieren nicht bei Bufotenin. Das Fehlen von mentalen halluzinogenen Wirkungen erlaubt dem User in hochdosierte, visionaere Stati zu gehen, ohne die Kontrolle zu verlieren. (dmt-nexus, Vlad, Dienstag, 7. Juli 2009 00:02:36)
Alexander Shulgin gibt die sehr niedere Dosis von 8 - 16 mg, i.v., in seinem Werk Tihkal an. (#101)
Ein posting zum Rauchen von Bufotenin-freebase:
In einer vor kurzem erschienenen Studie, durchgefuehrt von Jonathan Ott, testeten einige Personen die Wirkungen von gerauchter Bufotenin-freebase und alle Testpersonen berichtetn von starken visuellen Wirkungen, ohne die unangenehmen Nebenwirkungen vom Injizieren. Es ist staerker als DMT, aktiv bei Dosen von 2-10 mg. 10 mg ist eine riesige Dosis, welche eine voll ausgepraegten visionaeren Status erzeugt, und trotzdem fuehlst du dich ganz verbunden zur Realitaet. Es ist nicht ganz so freaky wie DMT sein kann. (dmt-nexus, Vlad, Dienstag, 7. Juli 2009 00:02:36)
Die Wirkdauer wird von A. Shulgin in seinem Werk "Tikhal" mit 1 - 2 h angegeben. (#101)
Das deckt sich auf mit unseren Beobachtungen mit den stark Bufotenin-haeltigen Anadenanthera colubrina-Art. Wir waren ungefaehr eine Stunde unterwegs nach Einsetzen der Wirkung. Mit der Reizschwelle und der Erwartungsphase am Anfang kommt man auf die 2 Stunden, die man benoetigt, um das Ganze abzuwickeln. Es ist ja noch die anfaengliche Uebelkeit mit Erbrechen zu ueberwinden, bis man in den Trip eingeht. Die Wirkung hoert fast schlagartig wieder auf, und man hat das Gefuehl aus einem Tagtraum aufzuwachen. Waehrend des Anandenanthera-Trips durch A. colubrina sieht man einen regelrechten Film mit geschlossenen Augen, den man sofort durch Augen-oeffnen unterbrechen kann. (eigen)
Ueber diese Substanz ist in der gesamten wissenschaftlichen Literatur kein Bericht ueber eine koerperliche oder psychische Abhaengigkeit erschienen. (eigen)
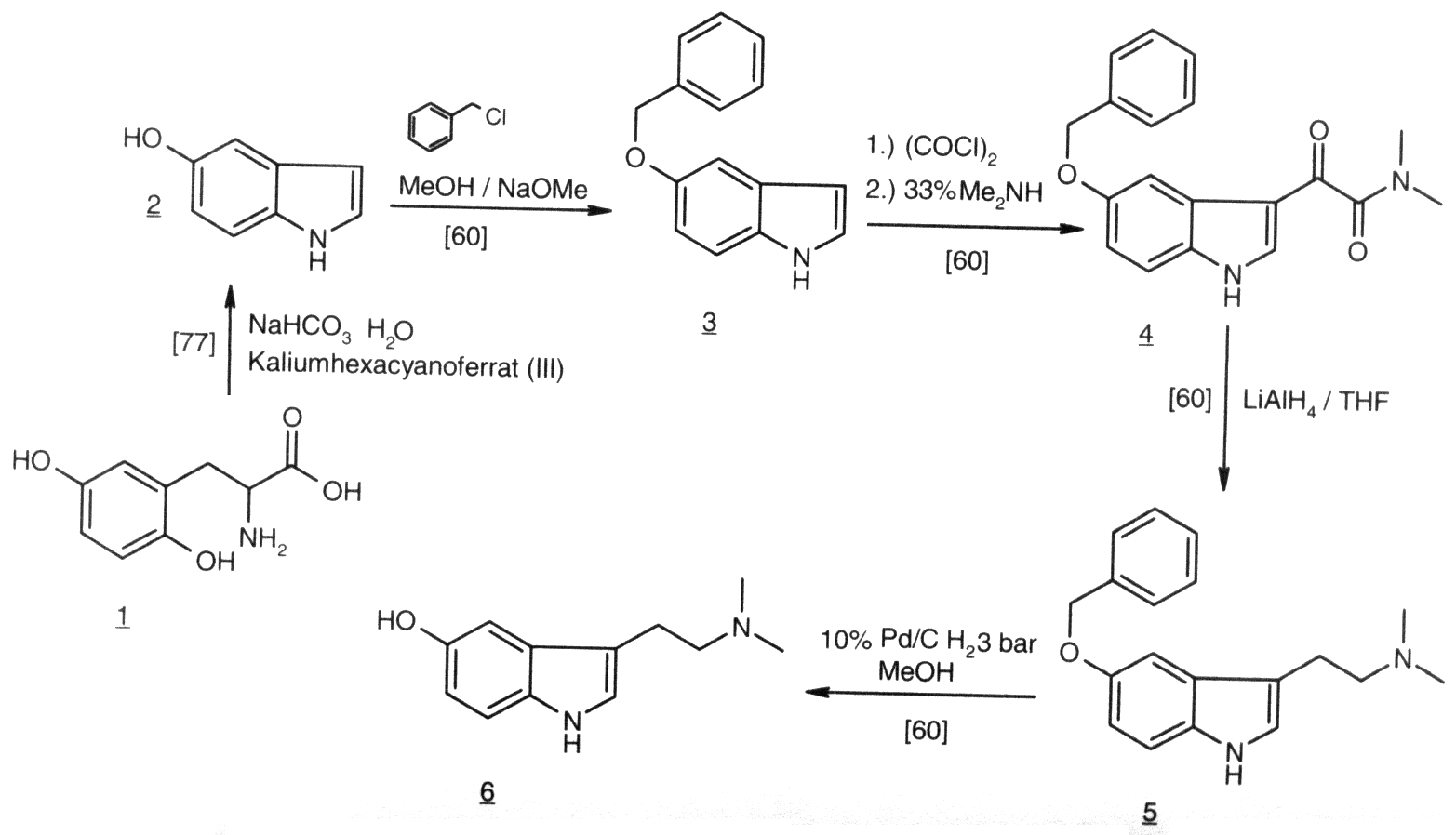
Auf DMT-Nexus wurde folgendes Verfahren unter dem Titel "No Smell, Quick and Easy, Almost Pure Bufotenine Extraction" in das Forum gepostet: (eigen)
Anbei wird folgende Extraktion, uebersetzt und kommentiert von mir, angegeben:
Ziel: Erhalte reine Bufotenin-freebase aus cebil Samen.
Materialien:
Prozedur:
1. Wiege und mahle die ganzen cebil (Anadenathera colubrina ) Samen zu einem feinen Pulver mit einer Kaffeemuehle oder einem elektrischen Haechsler, oder gar mit einem Moerser mit Stoessel.
2. Fuege rund 5g Natriumcarbonat hinzu und mixe, um eine gleichmaessige Mischung zu erhalten. (Klassisch waere, wenn man CaO oder Ca(OH)2 verwenden wuerde, so machen die Indianer die Reaktion, indem sie Asche verwenden und dazu noch Feuchtigkeit damit das CaO in Ca(OH)2 uebergeht, damit man damit free-basen kann; Anmerk. d. Verf.)
3. Fuege Wasser langsam hinzu, ein bisschen fuer ein bisschen, um eine teigartige Paste zu erzeugen. Bedecke den Kolben und lass es eine Stunde absitzen, um der Reaktion zu erlauben, fertig zu werden. Wir machen eine rohe Bufotenin-Freebase in diesem Schritt, welche Bufotenin beinhaltet, zusammen mit unerwuenschten, Uebelkeit produzierenden Komponenten, aus Pflanzenoelen und -fetten.
4. Wenn die richtige Konsistenz der Paste erreicht war im vorherigen Schritt, dann hat das Natriumcarbonat seine hydrierten Status gebildet und wir bleiben uebrig mit einem harten, trockenen Kuchen. Ein Geruch nach Ammoniak wird erscheinen, der uns anzeigt, dass sich die Freebase-Reaktion ereignet hat. Wenn ein Ueberschuss an Wasser gebraucht wurde, dann lass nur den Kolben unbedeckt und lass das Wasser verdunsten. Erhitzen und ein Ventilator wuerden diese Operation beschleunigen. Bufotenin ist sehr Hitze tolerant und das Wasser kann ruhig abgekocht werden, wenn du in Eile bist.
5. Mahle den harten Kuchen fein auf und dann bedecke ihn mit Wasserfreiem Aceton. Das Bufotenin wird sich im Aceton loesen, eine gelbe Loesung bildend, waehrend das ungebrauchte Natriumcarbonat zurueckgelassen wird. Giesse die Acetonloesung in einen Sammelkolben und wiederhole den Prozess durch Addition von mehr Aceton fur eine Gesamtmenge von 3 Extraktionen.
6. Filtriere die vereinten drei Aceton-Extrakte, durch einen Baumwollball, locker reingestopft in den verengenden Teil eines Trichters, um eine klar gelbe Acetonloesung der rohen Bufotenin-freebase zu erhalten.
7. Fuege den Inhalt einer 500 mg Fumarsaeure Kapsel direkt in diese Loesung und beobachte ein sofortiges Ausfallen des Bufoteninfumarats. Dies ist ein entfettender Schritt und beseitigt effektiv die Pflanzenoele und -fette. Lass das ganze Absitzen fuer eine Weile, um die Reaktion abzuschliessen. Das Bufoteninfumarat wird an den Seiten und am Boden des Kolbens picken und das verlorene Aceton, dass die unerwuenschten Oele beinhaltet, kann abgegossen werden und verworfen werden. Erlaube es den zurueckbleibenden Aceton zu verdunsten, bis der Geruch davon weg ist.
8. Fuege genug Wasser (rund 50 ml)hinzu, um das Bufoteninfumarat zu loesen und dann fuege rund ein Gramm Natriumcarbonat Pulver direkt zu dieser Loesung. Gleich wie Magie - bis sich ein karamell-gefaerbter Klumpen aus dem Freebase Bufotenin bilden wird. Dieser Klumpen sollte zusammenfallen, waehrend man einen Ueberschuss an Wasser hinzufuegt, um die komplette Penetration durch das Natriumcarbonat zu erlauben. Lass diese Reaktion kontinuerlich weitergehen, bis eine Weile vergangen ist zur Komplettierung, dann giesse das Wasser ab und trockne das karamell-farbene Bufotenin.
9. Loese komplett das unreine Bufotenin in 5 mL Aceton und fuege dann 20 ml Loesungsmittel( 1 Teil Aceton : 4 Teile Wundbenzin) dazu. Dies wird die unerwuenschten, Uebelkeit verursachenden, Verbindungen ausfaellen, wobei das reine Bufotenin in einer beinahe klaren Loesung zurueckbleibt. Erlaube der Reaktion genuegend Zeit um fertig zu werden.
10. Gebrauche den Medizintropfer (Pipette! Anmerk. d. Verf.), um die klare Bufotenin Loesung in einen anderen Kolben abzuheben, es bleiben die Uebelkeit verursachenden Laestigkeiten zurueck.
11. Verdampfe das Loesungsmittel, um einen hellgelben Niederschlag zu erhalten, wahrscheinlich reine Bufoteninfreebase. (www.dmt-nexus.com/Posted : Dienstag, 16. Februar 2010 18:26:57, by monkeyboy)
Eine zweite Extraktionsanleitung von Anadenanthera colubrina ausgehend, aber ohne Fumarsaeure, sondern mit Zitronensaeure und Calciumhydroxid:
Notiz: Um die Zitronensaeureloesung zu machen, loese 10 Gramm Zitronensaeure in 100ml Aceton. Es dauert etwas bis die Zitronensaeure sich im Aceton loest, also bereite diese Loesung schon einige Zeit vor der Extraktion.
Die unten angefuehrte Methode erfordert als Rohmaterial Bufotenin-Harz, welches aus Ethylacetat wenigstens einmal umkristallisiert worden ist:
1. Platziere das Bufoteninharz in ein Becherglas und fuege eine freizuegige Portion Feuerzeug-Benzin, Wundbenzin, Benzin, Waschbenzin dazu.
2. Erhitze das Becherglas auf einer heissen, elektrischen Platte und fuege Ethylacetat in kleinen Mengen aus einer kleinen Pipette hinzu, waehrend fortgesetzt geruehrt wird. Das Loesungsmittel sollte heiss sein, aber nicht sieden.
3. Fuege fortgesetzt Ethylacetat in kleinen Portionen hinzu, bis sich kein Harz mehr loest, und eine kleine Menge Restverunreinigen uebrigbleibt.
4. Entferne das Becherglas von der heissen Platte und lass es abkuehlen, bis die Verunreinigen sich abgesetzt haben, dann pipettiere die klar gelbliche Loesung in ein sauberes und kaltes Gefaess.
5. Wenn beim Abkuehlen die Loesung sehr milchig wird, fuege kleine Portionen von Ethylacetat hinzu, waehrend herumgewirbelt wird, bis es ist gerade schwach schimmert. Alternativ, wenn die Loesung klar bleibt beim Abkuehlen, fuege mehr Feuerzeug-Benzin tropfenweise hinzu, gleichzeitig herumwirbeln, bis eine durchgehende leicht schimmernde Farbe sich entwickelt. Sei vorsichtig, dass du nicht zuviel Feuerzeug-Benzin hinzufuegst, oder das Bufotenin wird sich in Klumpen niederschlagen. Wenn dies passiert ist, fuege mehr Ethylacetat hinzu und erhitze wieder, bis die Loesung wieder klar ist.
6. Bedecke das Becherglas mit einem Kreis aus Papiertaschentuechern, abgesichert mit einem Gummiband. Lass es an einen dunklen Platz ungestoert ruhen, bis das Loesungsmittel verdunstet ist. Typischerweise, beginnen sich die Kristalle zu Formen, rund um die Enden des fliehenden Loesungsmittels in Vorzug vor dem Formieren am Boden des Becherglases.
7. Wenn das Loesungsmittel komplett verdunstet ist, wurden die unreinen Kristallen herausgekratzt aus dem Becherglas, braune harzige Verunreinigungen zuruecklassend, welche dazuneigen sich am Boden zu bilden.
8. Die unreinen Kristalle koennen weiter gereinigt werden durch Umkristallisierung, die gleiche Methode wird oefters wiederholt. Wieauchimmer, weil die Kristalle leichter loeslich sind als die Verunrerinigungen, sollte das Becherglas herumgewirbelt werden nach jeder Addition Ethylacetat, eher als geruehrt werden. Die unloeslichen Verunreinigungen werden verworfen, wennn die Loesung durch eine Pipette in ein sauberes Becherglass transferiert wird. Adjustiere fuer das Schillern wie zuvor.
9. Die Umkristallisierung kann einige Male wiederholt werden, bis zur erwuenschten Reinheit. Typischerweise 3 oder 4 Umkristallisierungen sind genug, um kleine Haeufen weisser Nadeln aus reinem Bufotenin mit einem scharfen Schmelzpunkt von 126 Grad Celsius zu ergeben. (herbtalk.info, "Part 3. The crystallisation of bufotenine", stevek1, June 15, 2009, 04:53:16 AM)
Anbei zwei email-postings zum Thema Kristallisation von Bufotenin, die als Anleitung und Hinweis zur Erzeung von reinem, kristallisiertem Bufotenin dienen koennen:
Von Bufotenin wird berichtet, dass es eine Anzahl von unterschiedlichen Kristallformen hat, abhaengig von den Kristallisierungsbedinung und variierend mit aufeinanderfolgenden Umkristallisationen. In J. Ott (Bufotenine: Pharmanopo-Psychonautics, Journal of Psychoactive Drugs, 2001), sind drei Isomere beschrieben und charakterisiert worden durch ihre verschiedenen Schmelzpunkte. Keine Angabe wird gemacht zu den Kristallformen, anders beim Beschreiber: prismatisch fuer das Isomer, das bei 126 Grad Celsius schmolz. (herbtalk.info, "Part 3. The crystallisation of bufotenine", stevek1, June 15, 2009, 04:53:16 AM)
Bufotenin ist extrem schwer zu kristallisieren. Obwohl es loeslich ist in IPA, Aceton, DCM und Ethylacetat, fuehrt der Gebrauch von diesen Loesungsmitteln alleine unveraenderlich zu dem Ergebnis eines bernsteinfarbenen Harzes, anstelle der gewuenschten Kristalle. Wieauchimmer, der Gebrauch eines Alkan-Loesungsmittels in Verbindung mit Ethylacetat ergibt verlaessliche Ergebnisse, auch mit dem Harz Extrakt. Die besten Ergebnisse koennen erhalten werden durch den Gebrauch von Feuerzeug-Benzin (Ronsonol®), eher als Hexan als das Alkan der Wahl. Waehrend die erste Kristallisation dazuneigt kleine, amorphe, braeunliche Klumpen zu erzeugen, ergibt stufenweise Umkristallisation verschiedene Kristallformen und einen schrittweisen Wechsel der Farbe von gelb/braun zu farbloss/cremefarben. Die Kristallformen variieren von dndritisch (veraestelt), Baum-artigen Wachstum, ueber Nadelkissen aus stumpfen Nadeln, zu exquisiten kaputten Ventilatoren mit einem glaenzenden Schimmer. (herbtalk.info, "Part 3. The crystallisation of bufotenine", stevek1, June 15, 2009, 04:53:16 AM)
Alexander Shulgin beschreibt in seinem Buch Tihkal die Synthese dieser Verbindung:
Eine Loesung von 0.67 g 5-Hydroxyindol (Indol-5-ol) in 10 mL trockenem MeOH wurde mit einer Loesung von 0.30 g NaOMe in MeOH versetzt, gefolgt von 0.70 g Benzylchlorid. Die Mischung wurde auf dem Dampfbad 0.5 h lang erhitzt, und das Loesungsmittel unter Vakuum entfernt. Der Rest wurde suspensiert zwischen Wasser.htm">H2O und CH2Cl2, die organische Phase abgetrennt und die waessrige Phase wurde einmal mit CH2Cl2 extrahiert. Die vereinten Organika wurden vom Loesungsmittel unter Vakuum befreit, und der Rest wurde destilliert. Eine farblose Fraktion kam ueber bei 170-190 Grad Celsius und kristallisierte spontan im Empfaenger. Es wurden so 0.90 g (80%) 5-Benzyloxyindol mit einem Schmelzpunkt von 81-86 Grad Celsius erhalten, der beim Umkristallisieren aus Toluol/Hexan anstieg auf 94-96 Grad Celsius. Ueber ein Beispiel, bereitet durch die Decarboxylierung von 5-Benzyloxyindol-2-karbonsaeure, wurde berichtet, dass es einen Schmelzpunkt von 102 Grad Celsius aus Benzol hatte.
Eine Loesung von 1.0 g 5-Benzyloxyindol in 20 mL Et2O wurde auf 0 Grad Celsius abgekuehlt, kraeftig geruehrt, und mit 0.6 g Oxalylchlorid in 10 mL Et2O versetzt, tropfenweise hinzugefuegt, ueber die Dauer einer 0.5 h lang. Etwa am halben Weg der Addition erschien ein blass roter Feststoff. Das Ruehren wurde fuer zusaetliche 0,5h Stunden fortgesetzt und die Feststoffe wurde durch Filtration entfernt und mit einer kleinen Menge an Et2O gewaschen. Dieses Saeurechlorid hatte einen Schmelzpunkt von 149-151 Grad Celsius, wurde aber ohne weitere Reinigung oder Charakterisierung im naechsten Reaktionsschritt benutzt. Es wurde hinzugefuegt in kleinen Schritten zu 1.2 mL einer 33%-igen, waessrigen Loesung von Dimethylamin, verduennt mit angesaeuertem H2O, und die resultierenden Feststoffe wurden durch Filtration entfernt. Diese wurden mit H2O gewaschen, und dann mit Et2O und dann an der Luft getrocknet. Das Produkt, 5-Benzyloxy-N,N-dimethyl-3-indolglyoxylamid wog 1.18 g (82%) als es trocken war und hatte einen Schmelzpunkt von 185-187 Grad Celsius.
Zu einer gut geruehrten Suspension von 1.0 g LAH in 40 mL Et2O wurde eine Loesung von 1.0 g 5-Benzyloxy-N,N-dimethyl-3-indolglyoxamid in 15 mL THF hinzugefuegt. Als die Addition fertig war, wurde die Mischung 6h lang am Kochen am Rueckfluss gehalten, abgekuehlt, und das ueberschuessige Hydrid und der Reaktionskomplex vorsichtig zersetzt durch die Addition von H2O, und wenn die Wasserstoffentwicklung aufhoerte, wurde die Mischung basisch gemacht mit konzentrierter NH4OH. Die Feststoffe wurden durch Filtration entfernt und der Filterkuchen wurde mit THF gewaschen. Die Filtrate und Waschungen wurden vereint, und die Loesungsmittel wurden unter Vakuum entfernt, um einen klaren Rest zu ergeben, der in Et2O geloest wurde und angesaeuert wurde mit einer Loesung von Oxalsaeure in Et2O. Die gebildeten Kristalle wurden durch Filtration entfernt, mit Et2O gewaschen und an der Luft getrocknet, um 1.0 g (84%) 5-Benzyloxy-N,N-dimethyltryptaminoxalat mit einem Schmelzpunkt von 178-180 Grad Celsius nach dem Umkristallisieren aus MeOH zu ergeben. Das Hydrochloridsalz hatte einen berichteten Schmelzpunkt von 154-155 Grad Celsius und von 162-163 Grad Celsius.
Die Benzylgruppe wurde durch Hydrierung entfernt mit einer Loesung von 0.8 g 5-Benzyloxy-N,N-dimethyltryptaminoxalat in 5 mL MeOH, dass 0.1 g 10%-igen Pd/C Katalysator beinhaltete. Die Mischung wurde geschuettelt unter 3 atm Wasserstoff 6 h lang, und die Feststoffe wurden durch Filtration entfernt. Verdampfen des Loesungsmittels unter Vakuum ergab einen Rest, der in wasserfreiem Et2O geloest wurde und angesaeuert mit einer Loesung von Oxalsaeure in Et2O. Es wurden so erhalten, nach Filtration, Et2O Waschen und Lufttrocknen, 0.53 g (87%) Bufoteninmono-oxalat als pinke Nadeln, mit einem Schmelzpunkt von 93-94 Grad Celsius. Ein Schmelzpunkt von 178 Grad Celsius in der Literatur mag vom Bioxalat sein. Die freie Base hatte einen berichteten Schmelzpunkt von 125-126 Grad Celsius oder 146-147 Grad Celsius. (#101)
1934: Isolation von Bufotenin aus Kroeten: Wieland et al., Ann. 513, 1 (1934); (MerckIndex, 13. Aufl.)
1935: Eine Synthese von Bufotenin wurde veroeffentlicht: Hoshino, Shimodaira, Ann. 520, 19 (1935); (MerckIndex, 13. Aufl.)
1937: Isolation von Bufotenin aus Kroeten: Wieland, Wieland, ibid. 528, 239 (1937); (MerckIndex, 13. Aufl.)
1952: Eine Synthese von Bufotenin wurde veroeffentlicht: Harley-Mason, Jackson, Chem. & Ind. (London) 1952, 954; (MerckIndex, 13. Aufl.)
1953: Isolation von Bufotenin aus Giftpilzen: Wieland, Motzel, ibid. 581, 10 (1953). (MerckIndex, 13. Aufl.)
1954: Isolation von Bufotenin aus Piptadenia peregrina Benth. (jetzt Anadenanthera peregrina, Anmerk. d. Verf.), Leguminosae: Stromberg, J. Am. Chem. Soc. 76, 1707 (1954). (MerckIndex, 13. Aufl.)
1955: Eine Arbeit ueber Bufotenin ist erschienen: Stoll et al., Helv. Chim. Acta 38, 1452 (1955). (MerckIndex, 13. Aufl.)
1971: Die Bioaktivitaet von Bufotenin wurde in Indien erforscht: Bhattacharya, Sanyal, Indian J. Physiol. Pharmacol. 15, 133 (1971). (MerckIndex, 13. Aufl.)
1972: Die Kristall- und Molekularstruktur von Bufotenin wurde erforscht: G. Falkenberg, Acta Crystallogr. 28B, 3219 (1972). (MerckIndex, 13. Aufl.)
Abbildung 1: In: TRACHSEL Daniel, RICHARD Nicolas: "Psychedelische Chemie.", S. 364, Nachtschatten Verlag, Solothurn, 2000.
Das Quellenverzeichnis der Enzyklopaedie